Observable chez un nouveau-né sur 68 000 à 88 000, le syndrome d'Apert est dû à la mutation spécifique du gène FGFR2, qui a pour tâche de réguler la fusion des sutures crâniennes et le développement des doigts et des orteils.
Pour le diagnostic du syndrome d'Apert, un examen physique, une anamnèse, une évaluation radiologique du crâne et des doigts et des orteils et, enfin, un test génétique sont fondamentaux.
Actuellement, les personnes atteintes du syndrome d'Apert ne peuvent compter que sur des traitements symptomatiques, c'est-à-dire qu'ils soulagent les symptômes et évitent les complications les plus graves.
Brève revue des sutures crâniennes et de leur fusion
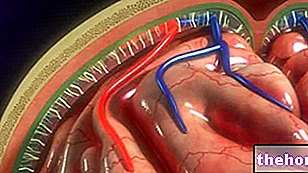
Les sutures crâniennes sont les articulations fibreuses, qui servent à fusionner les os de la voûte crânienne (c'est-à-dire les os frontaux, temporaux, pariétaux et occipitaux).
Dans des conditions normales, le processus de fusion des sutures crâniennes a lieu dans la période postnatale, commençant à 1-2 ans, pour certains éléments articulaires, et se terminant à 20 ans, pour d'autres. Ce processus de fusion long et cadencé permet au cerveau de grandir et de se développer adéquatement.
Le syndrome d'Apert doit cependant sa notoriété non seulement à son association avec la craniosténose, mais aussi au fait qu'il est lié à un certain degré de syndactylie, c'est-à-dire l'anomalie congénitale caractérisée par la fusion d'un ou plusieurs doigts ou doigts. pieds.
La possibilité de provoquer une craniosténose et une syndactylie en même temps fait du syndrome d'Apert un exemple d'acrocéphalosyndactylie ; en médecine, une « acrocéphalosyndactylie est une maladie génétique qui associe des malformations spécifiques du crâne (« acrocéphale » signifie « tête à pointe ») avec la fusion d'un ou plusieurs doigts ou orteils.
Quelles sont les conséquences de la fusion précoce de la suture crânienne ?
Si, comme dans le cas du syndrome d'Apert et d'autres maladies apparentées, la fusion des sutures crâniennes se produit au cours de la période prénatale, périnatale (*) ou de la très petite enfance, les organes cérébraux tels que le cerveau, le cervelet et le tronc cérébral, et le sens pendant que les yeux subissent altérations de la croissance et de la forme.
* N.B : la « vie périnatale » désigne la période comprise entre la 27e semaine de gestation et les 28 premiers jours suivant l'accouchement.
Épidémiologie : Quelle est la fréquence du syndrome d'Apert ?
Selon les statistiques, une personne sur 65 000 à 88 000 naît avec le syndrome d'Apert.
Saviez-vous que...
Les maladies génétiques qui, comme le syndrome d'Apert, provoquent une craniosténose sont environ 150.
Parmi ceux-ci, outre le syndrome d'Apert, le syndrome de Crouzon, le syndrome de Pfeiffer et le syndrome de Saethre-Chotzen se distinguent par leur importance.
Curiosité
La mutation acquise qui cause le syndrome d'Apert est un exemple de « mutation de novo», c'est-à-dire de « nouvelle mutation totalement dépourvue de caractère héréditaire ».
Quelles sont les causes de la mutation génétique associée au syndrome d'Apert ?
Prémisse: les gènes présents sur les chromosomes humains sont des séquences d'ADN qui ont pour tâche de produire des protéines fondamentales dans les processus biologiques essentiels à la vie, notamment la croissance et la réplication cellulaires.
Lorsqu'il est exempt de mutations (donc chez une personne saine), le gène FGFR2 impliqué dans le syndrome d'Apert produit dans les bonnes quantités une protéine réceptrice, appelée le Fibroblast Growth Factor Receptor 2, qui est essentielle pour marquer le timing de fusion du crâne. sutures et pour surveiller la séparation des doigts et des orteils (en d'autres termes, il signale le moment opportun pour la fusion des sutures crâniennes et régule la formation des doigts et des orteils).
En revanche, lorsqu'il subit la mutation observée en présence du syndrome d'Apert, le gène FGFR2 est hyperactif et produit la protéine réceptrice précitée en quantités si massives, que le timing relatif à la fusion des sutures crâniennes est altéré (il est plus rapide) et les processus de séparation des doigts et des orteils ne se déroulent pas correctement.
Qui est le plus à risque?
Concernant les cas acquis de syndrome d'Apert, les facteurs qui induisent la mutation du gène FGFR2 après la conception ne sont pas absolument clairs pour le moment.
Les recherches sur cet aspect sont toujours en cours.
Le syndrome d'Apert est une maladie autosomique dominante
Comprendre...
Chaque gène humain est présent en deux copies, appelées allèles, l'une d'origine maternelle et l'autre d'origine paternelle.
Le syndrome d'Apert présente toutes les caractéristiques d'une maladie autosomique dominante.
Une maladie génétique est autosomique dominante lorsque la mutation d'une seule copie du gène qui la provoque suffit à se manifester.
- Visage plat ou concave (en raison d'une croissance déficiente des os centraux du visage)
- Yeux gonflés, exorbités et grands ouverts orbites peu profondes et yeux anormalement espacés (hypertélorisme des orbites);
- Nez à bec;
- Mâchoire sous-développée, associée à un prognathisme ;
- Dents surpeuplées (en raison d'une mâchoire sous-développée)
- Oreilles plus basses que la normale.
Syndactylie
Chez les porteurs du syndrome d'Apert, la syndactylie est observée dans les mains, presque toujours, et dans les pieds, moins fréquemment que dans les mains.

Les caractéristiques typiques de la syndactylie entre les mains d'un individu atteint du syndrome d'Apert sont les suivantes :
- Présence d'un pouce court avec déviation radiale (c'est-à-dire orienté anormalement vers le radius, l'un des deux os de l'avant-bras) ;
- Syndactylie complexe entre l'index, le majeur et l'annulaire. Par syndactylie complexe, les médecins entendent une fusion anormale des doigts qui affecte non seulement les tissus mous (la peau), mais aussi les tissus osseux (les phalanges) ;
- Symphalangisme. C'est le terme médical qui désigne la fusion anormale des articulations interphalangiennes des doigts (les articulations interphalangiennes sont les éléments articulaires présents entre la phalange et la phalange) ;
- Syndactylie simple entre le quatrième et le cinquième orteil (c'est-à-dire entre l'annulaire et l'auriculaire) Avec la syndactylie simple, les experts se réfèrent à une fusion anormale des doigts qui n'affecte que les tissus mous (la peau).
GRAVITE DU SYNDROME DANS LE SYNDROME OUVERT : LES 3 TYPES
En fonction de la sévérité de la malformation du pouce (première des quatre caractéristiques), les experts du syndrome d'Apert distinguent 3 types de syndactylie de sévérité croissante :
- Le type I (le moins sévère) coïncide avec une « anomalie minime affectant le pouce, qui reste totalement indépendante de l'index ».
Autres anomalies: l'index, le majeur et l'annulaire sont fusionnés par une syndactylie complexe et présentent un symphalangisme affectant les articulations interphalangiennes distales ; c"est une syndactylie simple et incomplète entre l'annulaire et l'auriculaire (une syndactylie incomplète signifie que la fusion entre deux doigts est partielle).
Les autres informations: est le type le plus courant. - Le type II (gravité intermédiaire) se caractérise par une déviation radiale du pouce plus marquée, par rapport au cas précédent, et par un principe de syndactylie entre le pouce et l'index (c"est une syndactylie incomplète entre le pouce et l'index) .
Autres anomalies: index, majeur et annulaire sont les protagonistes d'une syndactylie complexe associée à un symphalangisme distal ; entre l'annulaire et l'auriculaire c"est une syndactylie simple et incomplète.
Les autres informations: c'est le deuxième type le plus courant. - Le type III (le plus sévère) est caractérisé par la présence d'un pouce uni en totalité à l'index, non seulement au niveau des tissus mous mais également au niveau des tissus osseux.
Autres anomalies: tous les doigts sont fusionnés, à tel point qu'il est presque impossible de les reconnaître ; c "est un" clou unique ; si entre les 4 premiers doigts la syndactylie est complexe, entre l'annulaire et l'auriculaire elle est (comme pour les autres types) simple et incomplète.
Les autres informations: c'est le type le plus rare.
Autres symptômes et signes possibles du syndrome d'Apert
Dans certains cas, en plus d'être associé à une craniosténose et à une syndactylie, le syndrome d'Apert est lié à la présence de : polydactylie (c'est-à-dire la présence d'un doigt supplémentaire dans les mains ou les pieds), perte auditive, récidive d'oreille et de sinus, hyperhidrose, peau, acné sévère, pas de poils sur les sourcils, fusion des vertèbres cervicales, syndrome d'apnées obstructives du sommeil et/ou fente palatine.
Complications
Les complications du syndrome d'Apert sont avant tout les conséquences graves que peut avoir la craniosténose sur le développement du cerveau et des capacités intellectuelles, et sur les capacités fonctionnelles des mains sujettes à la syndactylie.
Quand est-il possible de détecter le syndrome d'Apert ?
En règle générale, les anomalies crâniennes et numériques dues au syndrome d'Apert sont évidentes à la naissance, de sorte que le diagnostic et la planification du traitement sont immédiats.
à la tête (radiographies de la tête, scanner de la tête et/ou IRM de la tête) et des mains et éventuellement des pieds ; enfin, il se termine par un test génétique.
Examen physique et antécédents médicaux
L'examen physique et l'anamnèse consistent essentiellement en un examen précis des symptômes présentés par le patient.
Dans un contexte de syndrome d'Apert, c'est à ces moments de la démarche diagnostique que le médecin retrouve la craniosténose et la syndactylie, et leurs caractéristiques précises.
Examens radiologiques de la tête, des doigts et des orteils
Dans le cadre du syndrome d'Apert :
- Les examens radiologiques de la tête permettent au médecin de confirmer la présence d'une fusion précoce des sutures coronales (craniosynostose coronale ou brachycéphalie) ; de plus, elles lui permettent d'estimer la gravité des anomalies cranio-encéphaliques actuelles.
- En revanche, les examens radiologiques des doigts et des orteils sont indispensables non pas tant pour la confirmation de la syndactylie (pour cela l'examen visuel est suffisant), mais plutôt pour connaître en détail les caractéristiques des fusions interdigitales (type de syndactylie présente, niveau de fusion, etc.).
Test génétique
C'est l'analyse de l'ADN visant à détecter des mutations dans des gènes critiques.
Dans le cadre du syndrome d'Apert, il représente le test diagnostique de confirmation, car il met en évidence la mutation FGFR2 caractéristique de la maladie génétique en cause.
LES SOINS CHIRURGICAUX DE LA BRACHYCÉPHALIE
Pour le porteur du syndrome d'Apert, le traitement chirurgical de la brachycéphalie comprend :
- Une première intervention à un jeune âge (dans l'année de vie), visant à séparer plus tôt que prévu les sutures des fuses coronaires. Si cette intervention réussit, le cerveau bénéficie d'un bon espace de croissance et le risque de problèmes intellectuels est réduit.
- Une seconde intervention entre 4 et 12 ans, visant à redonner un aspect normal au visage, qui (le lecteur s'en souvient) est plat sinon concave.
L'opération en question implique l'incision de certains os du visage et leur repositionnement selon une disposition reflétant au moins partiellement la normalité. - Une troisième intervention éventuelle dans les années de l'enfance, dans le but d'éliminer ou au moins de réduire l'hypertélorisme oculaire.
LES SOINS CHIRURGICAUX DE SYNDACY
Le traitement chirurgical de la syndactylie varie selon les caractéristiques de la fusion interdigitée (il dépend donc du type).

Cela signifie que l'intervention valable pour un individu atteint du syndrome d'Apert peut ne pas être aussi valable pour un autre individu atteint de la même maladie génétique (elle n'est valable que si le type de syndactylie présente est le même).
Ayant clarifié cet aspect fondamental, le but de chaque type d'abord chirurgical existant est le même et consiste à libérer les doigts fusionnés, afin de garantir une certaine fonctionnalité aux mains.
Généralement, le traitement de la syndactylie comporte deux étapes :
- 1 étape : « libérer » le premier espace interdigital (espace entre le pouce et l'index) et le quatrième espace interdigital (espace entre l'annulaire et l'auriculaire) ;
- 2 étape : "libérer" le deuxième et le troisième espace interdigital (espace entre index et majeur, et espace entre majeur et annulaire).